


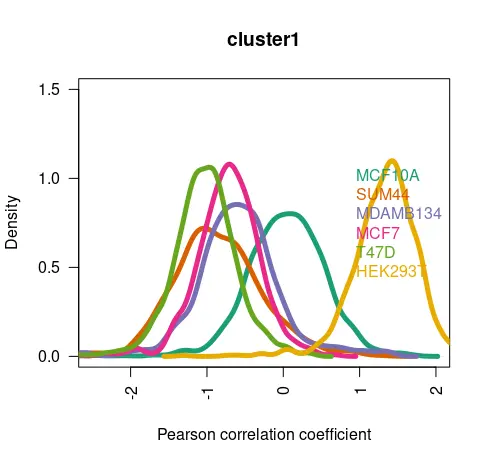
起因是因为看到了曾老师的一篇推文(https://mp.weixin.qq.com/s/S5TJ2JDOYAOynhjXwTH-FQ),其中是关于复现一篇cancer research《Single-cell transcriptomic heterogeneity in invasive ductal and lobular breast cancer cells》的细胞分群结果,见下图。
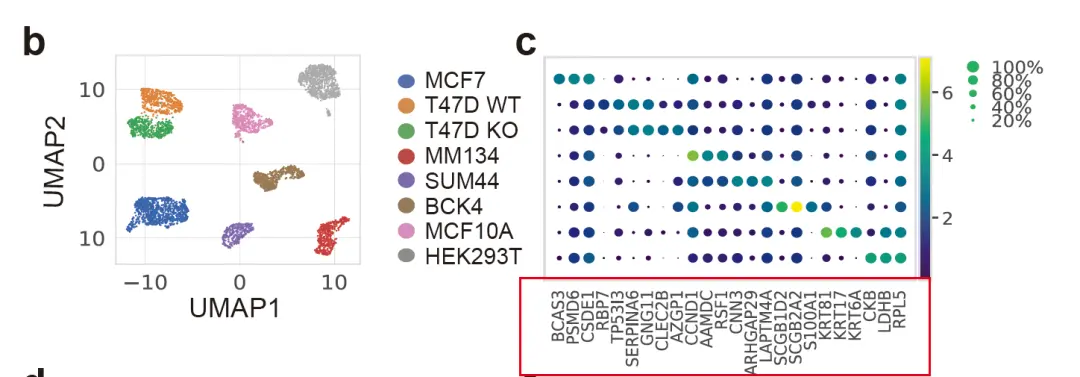

简单解释下图片内容,就是作者将MCF7,T47D,MM134等多个乳腺癌细胞系进行混合成一个样本进行单细胞测序。测序完之后,作者分出了8个细胞群,但这些细胞系之间的marker基因相互之间并不是那么的明显。作者这时候就有了一个特别棒的想法,就是去公共数据库找到相关细胞系的二代测序数据,然后将二代测序数据和单细胞数据进行相关性分析来确定分群结果,具体的复现结果可见我一开始提到的那篇推文。
觉得蛮有意思的,想着自己也跟着曾老师的代码一起复现下,结果看到了这句话

并且曾老师的推送里也没有附带他使用的表达矩阵,怎么办??只能自己提取呗
1、首先是作者使用的公共数据库的表达矩阵
找了好久,终于supplementary materials and methods找到相应数据来源。可以看到,MCF7,T47,MM134细胞均来自CCL数据库(SRR186687);SUM44,MCF10A来自GEO数据库(SRP026537,SRP064259),其中MCF10A在两个项目中都有,我选用的是SRP064259这个项目中的数据;HEK293T是直接下载了GEO数据库中的两个HEK293T的原始表达矩阵,然后取平均值即可。
(这边再说一个我踩坑的点,就是如果你想偷懒,直接去CCL和GEO下载他的raw data,你会发现这三个数据库的数据格式分别是FPKM,CPM和raw count格式,无法将其三者合并,所以只能老老实实的去下载他的fastq文件,自己跑一边上游提取表达矩阵)

我使用的fq文件是下面几个:
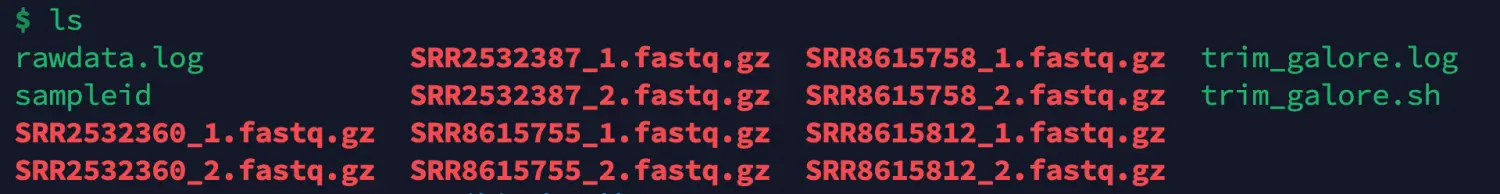
之后我们就进行常规流程的表达矩阵提取,首先是fastqc过滤,其中sample.ID文件里是我们的样本名字,如SRR2532360等,至于cleandata和rawdata的目录你们自己修改即可(以下的上游数据处理均由技能树的张娟老师提供)。下面的代码可以保存到脚本里面,使用nohup sh fastp.sh >fastp.log &挂载到后台运行,后同(其中在length这个参数中,我以往一般是设置36,但是我发现CCL数据库的和GEO数据库的read长度分别是102和52,所以根据正规流程,我们其实应该根据质控结果和经验分别设置他的length长度,对两个数据库的fastq数据分别过滤。但我这里只是为了复现,所以就直接使用默认的20)
cleandata=/home/project/data/cleandata/trim_galore
rawdata=/home/project/data/rawdata/
cat sample.ID | while read id
do
trim_galore --phred33 -q 20 --stringency 3 --fastqc --paired --max_n 3 -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz
done
接着是hisat2数据比对,我用的是公共数据,所以构建索引省略
index=refdir/server/reference/index/hisat2.2.1/hg38/genome
inputdir=/home/project/data/cleandata/trim_galore
outdir=/home/project/data/Mapping/Hisat2
cat /teach/t_rna/data/airway/sra/sample.ID | while read id
do
hisat2 -p 5 -x ${index} -1 ${inputdir}/${id}_1_val_1.fq.gz -2 ${inputdir}/${id}_2_val_2.fq.gz 2>${id}.log | samtools sort -@ 3 -o ${outdir}/${id}.Hisat_aln.sorted.bam - && samtools index ${outdir}/${id}.Hisat_aln.sorted.bam ${outdir}/${id}.Hisat_aln.sorted.bam.bai
done
最后就是featurecount进行表达定量
gtf=/home/data/refdir/server/reference/gtf/ensembl/Homo_sapiens.GRCh38.98.gtf.gz
inputdir=/home/project/data/Mapping/Hisat2
# featureCounts对bam文件进行计数
featureCounts -T 6 -p -t exon -g gene_id -a $gtf -o all.id.txt $inputdir/*.sorted.bam
# 得到表达矩阵
cat all.id.txt | cut -f1,7- > counts.txt
# 处理表头部分省略,大家自己根据需要使用sed处理即可
以上,我们获得了作者的公共数据库raw count文件(上游过程不好解释,初学的小伙伴记得多去找些资料理解下)
2、接下来就简单了,就是对原始表达矩阵的处理和单细胞数据的处理
library(stringr)
library(Seurat)
#1.首先处理单细胞数据,获得各细胞群的平均表达矩阵
{
#以下所用的文件去文章对应的GSE144320下载即可,作者的表达矩阵颠倒了
counts=read.csv(file = "input/GSM4285803_scRNA_RawCounts.csv",row.names = 1)
counts=as.matrix(counts)
counts=t(counts)
counts=as.data.frame(counts)
meta=read.csv(file="input/GSM4285803_scRNA_metaInfo.csv",row.names = 1)
identical(colnames(counts),rownames(meta))
#获得了表达矩阵和患者信息,构建seurat对象
sce 接下来是处理GEO和CCL的公共数据
#2.处理作者使用的CCL和GEO上的公共数据表达矩阵
{
rawcount1=read.table("input/rawcounts.txt",row.names = 1,sep = "t",header = T)
rawcount2=read.table("input/GSM1867011_HEK_293_C1_USC_RNA_40.csv",row.names = 1,sep = "t",header = T)
rawcount3=read.table("input/GSM1867012_HEK_293_C2_USC_RNA_41.csv",row.names = 1,sep = "t",header = T)
identical(rownames(rawcount2),rownames(rawcount3))
rawcount23=cbind(rawcount2,rawcount3)
colnames(rawcount23)=c("GSM1867011","GSM1867012")
rownames(rawcount23)=str_sub(rownames(rawcount23),1,15)#这个需要提前确定,.号前面的ENSG编号没有重复
rawcount23=as.data.frame(rowSums(rawcount23)/2)
colnames(rawcount23)="HEK293T";colnames(rawcount1)=c("MCF10A","SUM44","MDAMB134","MCF7","T47D")
#以上是rawcount矩阵,现在转化为cpm表达矩阵
#扫了一遍发现,HEK293T的rawcount下载下来后数值都很高,而且没有0值,猜测作者在上传前就已经对原始矩阵进行过过滤
#既然如此,我们就不对我们的表达矩阵进行过滤,与HEK293T的rawcount进行合并即可
library(edgeR)
tmp=intersect(rownames(rawcount1),rownames(rawcount23))
length(tmp);dim(rawcount1);dim(rawcount23)
#获得最后的合并后的表达矩阵,进行cpm转换,需要log,不然差异过大,见https://cloud.tencent.com/developer/article/1625229
rawcount=cbind(rawcount1[tmp,],rawcount23[tmp,])
colnames(rawcount)[6]="HEK293T"
cpmcount=log2(cpm(rawcount)+1)
#进行基因id的转换
library(clusterProfiler)
library(org.Hs.eg.db)
id2symbol 3.其中av和cpmcount我们都获得了,接下来就开始相关性表达矩阵的分析
{
#首先是作者已经定义好的细胞群
load(file="Rdata/section8.Rdata")
head(av)
av=as.data.frame(av);cpmcount=as.data.frame(cpmcount)
dim(av);dim(cpmcount)
av=av[intersect(rownames(av),rownames(cpmcount)),]
cpmcount=cpmcount[intersect(rownames(av),rownames(cpmcount)),]
dim(av);dim(cpmcount)
data_merged=cbind(av,cpmcount)
tt 在进行相关性分析后,我发现,我使用cpm数据做出来的数据并没有曾老师的热图显著,很明显用我的热图去进行细胞群的划分十分牵强。我猜测可能是上游数据中,我和曾老师在数据处理中的参数设置问题,但我没有曾老师的原始代码,现在也无从考证。
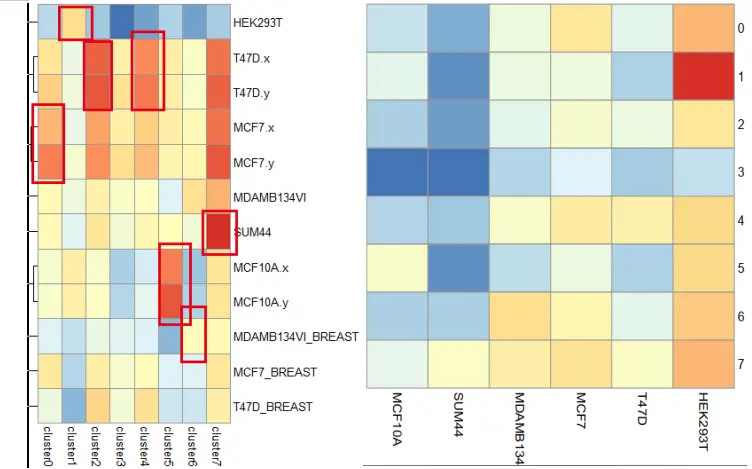
那现在属于复现成功了吗??我突然想到,既然我们的根本目的只是为了将其分群,那我们只需要cluster_x群细胞在某个种类细胞系中的相关性结果即可。即当我们去计算总体的相关性时,cluster之间时会相互影响的。所以,我们对结果进行scale处理。
pheatmap::pheatmap(tt1,cluster_rows = F,cluster_cols = F,scale = "column")
#这里我确实不知道该怎么解释,请大家自行理解下
之后,就得到和作者以及曾老师一模一样的相关性热图了;;可见数据总体的表达还是没有问题的,可能就是上游的一些参数处理问题
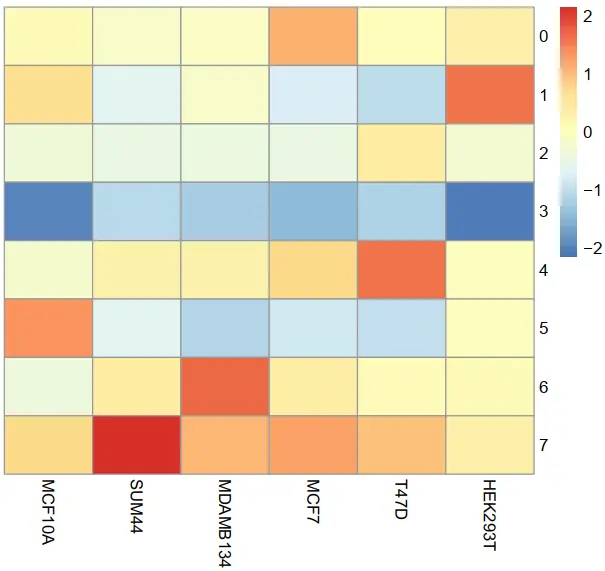
复现作者的图(具体代码就不贴了,具体可见曾老师写的推文《如果你觉得相关性热图不好看,或者太简陋》)
!!!!需要注意的是,在你获得相关性矩阵后,在画图前,也要和上面一样scale一下
写在文末的几句话:花了好几天的时间磕磕碰碰的把曾老师的这篇推文给复现出来,花费时间最长的竟然还是在获得公共数据库表达矩阵那里。我整篇推文基本把二代测序的上游数据处理和seurat结构给简单的回顾了一遍,包括我在找这些数据时候遇到的各种坑。授人以鱼不如授人以渔,多敲代码多犯错,才能更好的进步。
药物型尖端扭转型室性心动过速:一个白天上手术,晚上写代码的苦逼外科医生φ(..)
【信息由网络或者个人提供,如有涉及版权请联系COOY资源网邮箱处理】






暂无评论内容